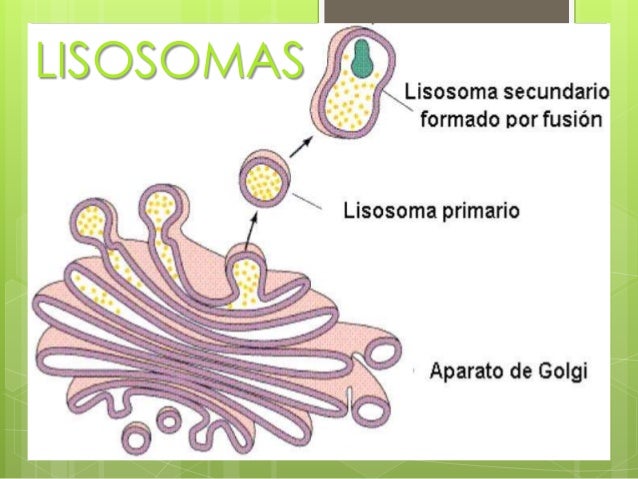
¿QUE SON Y DONDE SE ENCUENTRAN?
- Son orgánulos relativamente grandes, formados por el Aparato de Golgi, que contienen enzimas hidrolíticas y proteolíticas encargadas de degradar material intracelular de origen externo (heterofagia) o interno (autofagia) que llegan a ellos. Es decir, se encargan de la digestión celular. Son estructuras esféricas rodeadas de membrana simple. Son bolsas de enzimas que si se liberasen, destruirían toda la célula. Esto implica que la membrana lisosómica debe estar protegida de estas enzimas. El tamaño de un lisosoma varía entre 0,1-1,2 μm. Los lisosomas fueron descubiertas por el bioquímico belga Christian de Duve en 1974.
- En un principio se pensó que los lisosomas serían iguales en todas las células, pero se descubrió que tanto sus dimensiones como su contenido son muy variables. Se encuentran en todas las células animales y vegetales.
CLASIFICACION:
Lisosomas primarios: son pequeños y con escasas enzimas lisosómicas. Estos son los que se producen directamente en el aparato de Golgi, contienen hidrolasas ácidas. Cuando la clatrina (que es la proteina que las recubre), quedan sueltos con sus enzimas lisosómicas y aún no han degradado material celular.
Lisosomas secundarios: son grandes e irregulares y forman los endosomas que permiten la autofagia de las organelas numerosas o viejas.
Endosomas: conjunto de lisosomas secundarios con acción de autofagia.
Cuerpos multivesiculares: se forman por la fusión entre los endocomas tardíos y tempranos, contienen abundantes enzimas endocíticas.
Fagolisosomas: se forman por la fusión de una vacuola fagocítica con endosoma tardío o lisosoma.
Autofagolisosomas: fusión de una vacuola autofágica con un endosoma tardío o un lisosoma.
Cuerpos residuales: lisosomas sin capacidad para degradar material o contienen material no degradable.
ENFERMEDADES LISOSOMICAS:
- En las enfermedades de almacenamiento lisosómico, alguna enzima del lisosoma tiene actividad reducida o nula debido a un error genético y el substrato de dicho enzima se acumula y deposita dentro del lisosoma que aumentan de tamaño a causa del material sin digerir, lo cual interfiere con los procesos celulares normales; algunas de estas enfermedades son:
- Son enfermedades causada por la disfunción de alguno de los enzimas de la ruta de degradación de los esfingolípidos.
- La lipasa ácida es una enzima fundamental en el metabolismo de los triglicéridos y del colesterol, que se acumulan en los tejidos. La disfunción de esta enzima provoca dos enfermedades, la enfermedad de almacenamiento de ésteres de colesterol, en que la enzima presenta muy poca actividad, y la enfermedad de Wolman, en que la enzima es totalmente inactiva.
Glucogenosis tipo II o enfermedad de Pompe.
Mucopolisacaridosis.
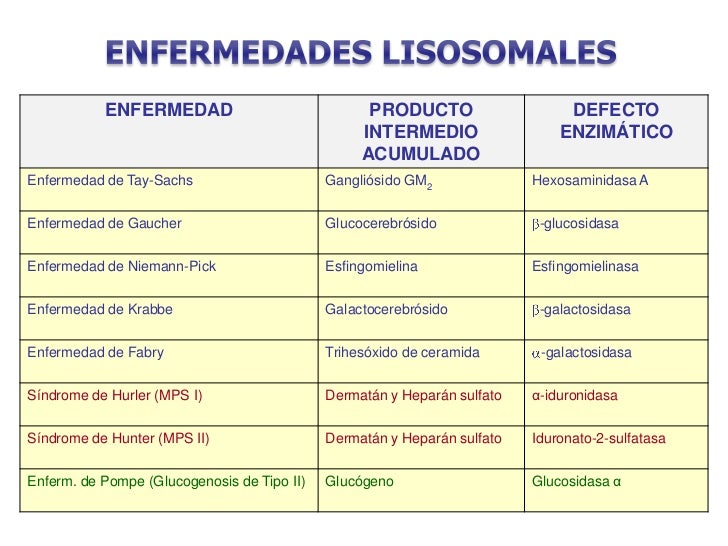
- Es un defecto de la α(1-4) glucosidasa ácida lisosómica, también denominada maltasa ácida, lo que causa la acumulación de glucógeno en los lisosomas. En niños destaca por producir insuficiencia cardíaca al acumularse en el músculo cardíaco causando cardiomegalia. En adultos el acúmulo es más acusado en músculo esquelético.
- Causadas por la ausencia o el mal funcionamiento de las enzimas necesarias para la degradación moléculas llamadas glicosoaminoglicanos o glucosaminglucanos (antes llamadas mucopolisacáridos), que son cadenas largas de hidratos de carbono presentes en todas las células que intervienen en la construcción de los huesos, cartílagos, tendones, córneas, la piel y el tejido conectivo.










No hay comentarios.:
Publicar un comentario